Homocystinuria: A treatable disorder
The Homocystinurias are a group of inherited metabolic disorders leading to accumulation of homocysteine and its metabolites in the blood and urine. The most common of these disorders is known as classical homocystinuria (HCU) and is caused by a deficiency in the enzyme known as cystathionine beta-synthase or CBS. There are other very rare metabolic causes of homocystinuria that are quite different from HCU. The other forms of homocystinuria are not discussed in this checklist.
The reported international incidence of classical HCU varies from 1 in 50,000 to 1 in 200,000[1]. The true prevalence, especially in more mild cases is unknown.
In the United States homocystinuria is included in the disorders screened at birth by newborn screening. Screening does not detect all cases of homocystinuria. If not diagnosed by newborn screen, delays are on average 4.5 years from disease onset to accurate diagnosis[2].
Could it be classical homocystinuria?
HCU is present at birth; the symptoms and signs may develop in infancy, childhood, or even in adulthood[3]. The symptoms of classical homocystinuria vary greatly, with some affected individuals only having mild signs of the disorder; others may have many different symptoms, including some potentially life-threatening complications[4]. The most commonly affected areas are the brain, the eyes, the skeleton, and the vascular system. Due to the symptoms and signs being nonspecific, this often leads to misdiagnosis and late diagnosis[3,5].
Key Warning signs and symptoms include:
It is important to note that early treatment can prevent the development of the symptoms listed above or prevent further complications and worsening of already present symptoms [3]. Speak with your doctor about your child’s symptoms and take this HCU Disease Checklist along to your appointment.
Genetic Inheritance
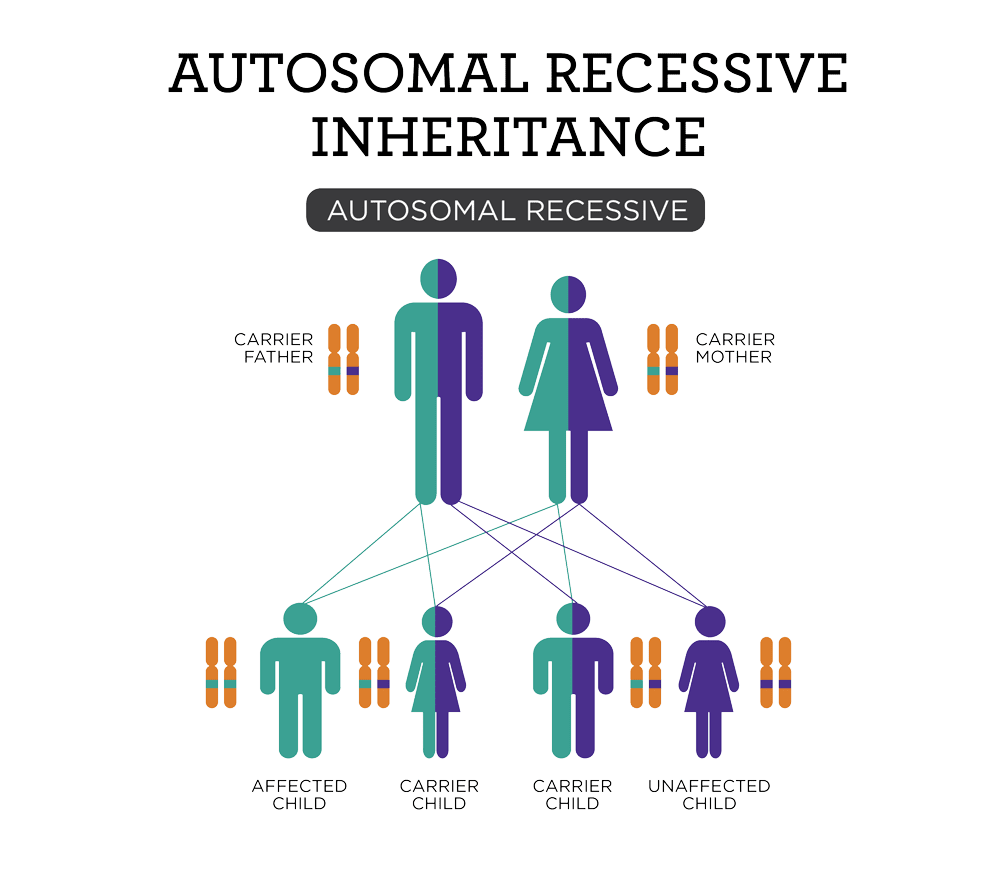
It is suggested that relatives of a family member recently diagnosed with homocystinuria should be tested[5]. Homocystinuria is an autosomal recessively inherited disorder. This means that an individual inherits the same defective copy of the CBS gene from each parent. The risk is the same for males and females.
What to do if there is a suspicion of HCU?
If an individual is showing any of the signs above then classical homocystinuria should be considered as a potential diagnosis. It is important to measure the plasma total homocysteine level and if high (above 50 µM) and homocystinuria is suspected refer patient to a metabolic center for further investigation and early treatment where appropriate[1] .
Please visit our Find a Clinic feature to find a clinic that can test you.
Classical homocystinuria is a treatable disease, and early diagnosis and treatment can prevent the development or progression of the complications associated with the disease. If untreated, classical homocystinuria is a serious and potentially fatal disease. Timely diagnosis and treatment of classical homocystinuria is important in preventing or reducing the symptoms associated with the disorder[1] . Early diagnosis and treatment can make a real difference in patient outcomes[6] .
HCU Network America
For more information about classical homocystinuria or patient support, please visit: hcunetworkamerica.org
If you have any questions for us, please send us an email at info@hcunetworkamerica.org
This information has been used with permission from HCU Network Australia
References:
[1]Medscape. http://emedicine.medscape.com/article/1952251-overview.
[2]HCU Network Australia, unpublished data.
[3]García-Jiménez MC, Baldellou A, García-Silva MT, Dalmau-Serra J, Garcia-Cazorla A, Gomez-Lopez L, Pedron Giner C, Alonso Luengo O, Pena Quintana L, Luz Couce M, Martinez-Pardo M, Lambruschini N (2012), Epidemiological study of the metabolic diseases with homocystinuria in Spain. Anales de Pediatria, 76, 133-139
[4]Morris, A.A.M., Kožich, V., Santra, S (2016), Guidelines for the diagnosis and management of cystathionine beta-synthase deficieny, J Inherit Metab Dis, 40, 49-74
[5]Refsum H, Smith AD, Ueland PM, Nexo E, Clarke R, McPartin J, Johnston C, Engbaek F, Scheede J, McPartin C, Scott JM (2004), Facts and recommendations about total homocysteine determinations: an expert opinion, Clinical Chemisty, 50, 3-32
[6]Schiff M, Blom HJ (2012), Treatment of inherited homocystinurias. Neuropediatrics 43, 295-304
Doesn't sound like HCU but still concerned?
If your child has these symptoms, it could still be genetic! We recommend you find a genetic counselor in your area and have your child tested. Please visit the National Society for Genetic Counselors to find a genetic counselor near you.
